
Major components
Here we detail major and well-studied basement membrane components and highlight their links to human disease.
On this page:
Type IV collagen
Laminins
Heparan-sulfate proteoglycans
Type XVIII collagen
Nidogens/entactins
Type IV collagen
A large disulfide-bonded trimeric protein ubiquitous to all basement membranes. It is formed by a stoichiometric combination of six different chains (α1 to α6) encoded by different genes (COL4A1 to COL4A6). These different chains assemble intracellularly into 400 nm-long rod-like triple helical heterotrimers (α1α2α1, α3α4α5 and α5α6α5). They are subsequently secreted to the extracellular environment to form the central collagenous core of all basement membranes, providing them with long-term structural and mechanical stability.
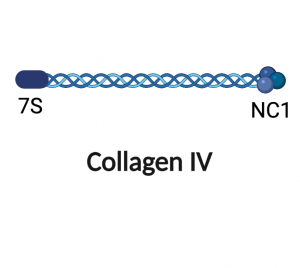
Type IV collagen α1/α2 chains
Multi-system disorder caused by variants in the autosomal COL4A1 and COL4A2 genes and characterised by abnormal blood vessels in multiple sites (brain, eyes, muscle, kidney). Due to the widespread distribution of type IV collagen α1 and α2 chains, variants in their encoding genes often lead to systemic complications associated with failures in basement membrane assembly in many tissues such as brain, kidneys and lungs, however, the full spectrum of abnormalities is still not well characterised. Related disorders include autosomal porencephaly, schizencephaly, HANAC syndrome (angiopathy with haematuric nephropathy, aneurysms, ophthalmological complications and muscle cramps), retinal arterial tortuosity, brain small vessel disorders and increased susceptibility for aneurysms and intracerebral haemorrhage.
See clinical phenotypes for more information
Papers
- COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. Plaisier, E et al. N Engl J Med. 2007 Dec 21. PMID: 18160688
- COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Jeanne, M et al. Am J Hum Genet. 2012 Jan 13. PMID: 22209247
- HANAC Syndrome Col4a1 Mutation Causes Neonate Glomerular Hyperpermeability and Adult Glomerulocystic Kidney Disease. Chen, Z et al. J Am Soc Nephrol. 2016 Apr 27. PMID: 26260163
- Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Plaisier, E et al. Am J Med Genet A. 2010 Oct 15. PMID: 20818663
- Role of COL4A1 in small-vessel disease and hemorrhagic stroke. Gould, DB et al. N Engl J Med. 2006 Apr 6. PMID: 16598045
- COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Lanfranconi, S and Markus, HS. Stroke. 2010 Aug. PMID: 20558831
- Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Poschl, E et al. Development. 2004 Apr. PMID: 14998921
Alport syndrome (type IV collagen α3/α4/α5 chains)
The most common hereditary glomerular disease in humans caused by variants in the COL4A5 gene on chromosome Xq22.3 (X-linked disease; accounts for 80% of the cases) or in the autosomal COL4A3 or COL4A4 genes on chromosome 2q36.3 (autosomal recessive disease; 10% of the cases). This disease affects the basement membranes in the kidneys, lungs, ears and eyes. In the kidney, the pathognomonic alteration is a thick, lamellated, “basket-weave” glomerular basement membrane due to the loss of type IV collagen α3α4α5 trimers.
See clinical phenotypes for more information
Papers
- Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Barker, DF et al. Science. 1990 Jun 8. PMID: 2349482
- Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Mochizuki, T et al. Nat Genet. 1994 Sept. PMID: 7987396
- A review of clinical characteristics and genetic backgrounds in Alport syndrome. Nozu, K et al. Clin Exp Nephrol. 2019 Feb. PMID: 30128941
- Basement Membrane Defects in Genetic Kidney Diseases. Chew, C and Lennon, R. Front Pediatr. 2018 Jan 29. PMID: 29435440
- Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Cosgrove, D et al. Genes Dev. 1996 Dec 1. PMID: 8956999
- Mouse model of X-linked Alport syndrome. Rheault, MN et al. J Am Soc Nephrol. 2004 Jun. PMID: 15153557
Anti-glomerular basement membrane disease and Goodpasture syndrome (type IV collagen, predominantly the α3 chain)
Autoimmune diseases caused by the abnormal production of autoantibodies against epitopes on type IV present in the glomerular and alveolar basement membranes. This leads to crescentic glomerulonephritis (anti-GBM disease) that can be associated with life-threatening lung haemorrhage (Goodpasture syndrome).
See clinical phenotypes for more information
Papers
- Anti-Glomerular Basement Membrane Disease. McAdoo, SP and Pusey, CD. Clin J Am Soc Nephrol. 2017 Jul 7. PMID: 28515156
- Goodpasture’s autoimmune disease – A collagen IV disorder. Pedchenko, V et al. Matrix Biol. 2018 Oct. PMID: 29763670
- Basement Membrane Defects in Genetic Kidney Diseases. Chew, C and Lennon, R. Front Pediatr. PMID: 29435440
Laminins
The major non-collagenous heterotrimeric glycoprotein found in the basement membranes which are critical for many cellular processes. The laminin protomer (approx. 500 kDa) is assembled intracellularly as a disulfide-bonded, cruciform, heterotrimer. It is formed by three genetically distinct chains (α,β,γ), with three short arms and a long arm formed by the α-helical arrangement of the coiled-coil domains. So far, the genes for 5 α chains, 4 β chains and 3 γ chains have been identified and although 50 heteromeric combinations are theoretically possible, only 16 have been suggested to naturally occur.
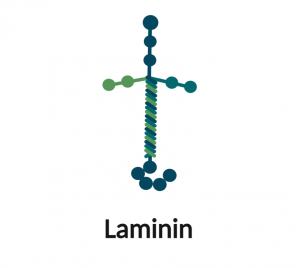
Junctional epidermolysis bullosa (Laminin-332)
A genetic disease caused by variants in the genes encoding the three subunits of laminin-332 (LAMA3, LAMB3 and LAMC2). It is characterised by diminished epidermal-dermal adhesion that leads to fragile skin, mechanically inducible blisters on the skin and mucosal membranes, and a series of extracutaneous manifestations and complications.
See clinical phenotypes for more information
Papers
- Laminin 332 in junctional epidermolysis bullosa. Kiritsi, D et al. Cell Adh Migr. 2013 Jan-Feb. PMID: 23076207
- Hereditary epidermolysis bullosa. Laimer, M et al. J Dtsch Dermatol Ges. PMID: 26513070
- Gentamicin induces LAMB3 nonsense mutation readthrough and restores functional laminin 332 in junctional epidermolysis bullosa. Lincoln, V et al. Proc Natl Acad Sci U S A. 2018 Jul 10. PMID: 29946029
Poretti–Boltshauser syndrome (Laminin α1 chain)
Autosomal recessive cerebellar dysplasia and cysts which can be associated with eye abnormalities (myopia, retinal dystrophy and ocular motor apraxia). It is caused by homozygous or compound heterozygous mutations in the LAMA1 gene. Patients can also present with delayed motor and language development, and intellectual disability.
See clinical phenotypes for more information
Papers
- Ataxia, intellectual disability, and ocular apraxia with cerebellar cysts: a new disease?. Poretti, A et al. Cerebellum. 2014 Feb. PMID: 24013853
- Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. Aldinger, KA et al. Am J Hum Genet. 2014 Aug 7. PMID: 25105227
- Clinical, neuroradiological and molecular characterization of cerebellar dysplasia with cysts (Poretti–Boltshauser syndrome). Micalizzi, A et al. Eur J Hum Genet. 2016 Aug. PMID: 26932191
Pierson syndrome (Laminin ß2 chain)
This rare disease is also known as microcoria-congenital nephrotic syndrome, and it is caused by homozygous or compound heterozygous variants in the LAMB2 gene that encodes the laminin ß2 chain. Individuals affected lack this laminin ß chain in the basement membranes of the kidney as well as other sites and develop distinct ocular abnormalities and congenital nephrotic syndrome within the first year of life.
See clinical phenotypes for more information
Papers
- Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Zenker, M et al. Hum Mol Genet. 2004 Nov 1. PMID: 15367484
- Congenital nephrosis, mesangial sclerosis, and distinct eye abnormalities with microcoria: an autosomal recessive syndrome. Zenker, M et al. Am J Med Genet A. 2004 Oct 1. PMID: 15372515
- Alport syndrome and Pierson syndrome: Diseases of the glomerular basement membrane. Funk, SD et al. Matrix Biol. 2018 Oct. PMID: 29673759
- Proteinuria precedes podocyte abnormalities inLamb2-/- mice, implicating the glomerular basement membrane as an albumin barrier. Jarad, G et al. J Clin Invest. 2006 Aug. PMID: 16886065
Merosin-deficient congenital muscular dystrophy (Laminin α2 chain)
Caused by autosomal recessive variants in the LAMA2 gene encoding the α2 subunit of laminin-221. This results in diminished or abolished expression of this protein and destabilisation of basement membranes in skeletal muscle and brain tissues. Main symptoms include mild to severe early-onset muscular dystrophy (hypotonia, muscle weakness and wasting), somatosensory abnormalities, respiratory dysfunction and epilepsy.
See clinical phenotypes for more information
Papers
- Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Yurchenco, PD et al. Matrix Biol. 2018 Oct. PMID: 29191403
- Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Reinhard, JR et al. Sci Transl Med. 2017 Jun 28. PMID: 28659438
- Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. Kuang, W et al. J Clin Invest. 1998 Aug 15. PMID: 9710454
- Laminin alpha2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Guo, LT et al. Neuromuscul Disord. 2003 Mar. PMID: 12609502
- Merosin/laminin-2 and muscular dystrophy. Wewer, UM and Engvall, E. Neuromuscul Disord. 1996 Dec. PMID: 9027848
Heparan-sulfate proteoglycans
The main heparan-sulfate proteoglycans (HSPG) found in basement membranes are perlecan and agrin. Perlecan is an integral proteoglycan of basement membranes ubiquitously expressed throughout the body. It contains a large multiple-domain protein core (approx. 470 kDa) with binding sites for 3 or 4 heparan-sulphate chains and several O-linked oligosaccharides. Perlecan is critical for branching morphogenesis during development, angiogenesis, basement membrane assembly and establishment of its charge profile, neuromuscular function, cartilage formation and others.
Agrin is found in specialised basement membranes in the kidneys, lungs, small blood vessels and neuromuscular junctions. It is formed by a large protein core (approx. 220 kDa) that carries at least two heparan or chondroitin-sulfate lateral chains, binding sites for basement membrane proteins (laminins and collagen IV) and cell receptors (α-distroglycan and integrins). In mice, there are two different isoforms with distinct N-termini (short and long N-termini) and differential subcellular and tissue expression.

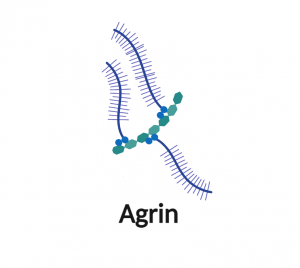
Schwartz Jampel syndrome type-1 (perlecan)
Rare autosomal recessive musculoskeletal disease caused by hypomorphic variants in the HSPG2 gene (encoding perlecan). It is characterised by skeletal muscle abnormalities (permanent myotonia), growth defects, chondrodystrophy and chondrodysplasia, and ocular and facial abnormalities that usually become apparent during childhood. Neuromyotomia is associated with congenital peripheral nerve hyperexcitability.
See clinical phenotypes for more information
Papers
- Schwartz-Jampel syndrome and perlecan deficiency. Stum, M et al. Acta Myol. 2005 Oct. PMID: 16550923
- Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nicole, S et al. Nat Genet. 2000 Dec. PMID: 11101850
- A mouse model of Schwartz-Jampel syndrome reveals myelinating Schwann cell dysfunction with persistent axonal depolarization in vitro and distal peripheral nerve hyperexcitability when perlecan is lacking. Bangratz, M et al. Am J Pathol. 2012 May. PMID: 22449950
- Reduced perlecan in mice results in chondrodysplasia resembling Schwartz-Jampel syndrome. Rodgers, KD et al. Hum Mol Genet. 2007 Mar 1. PMID: 17213231
Nidogens/entactins
Dumbbell-shaped molecules with three globular domains (G1, G2 and G3) separated by two strand domains, with anchoring binding sites to other basement membrane core components such as type IV collagen, laminins and heparan-sulfate proteoglycans. There are two genetically distinct homologue isoforms (nidogen-1/entactin-1 with 150 kDa, and nidogen-2/entactin-2 with 200 kDa) that share domain arrangement similarities.
Dandy-Walker malformation and occipital cephaloceles
Variants in NID1 encoding the protein nidogen-1 have been implicated in the pathogenesis of the autosomal dominant Dandy-Walker malformation and occipital cephaloceles.
Papers
- NID1 variant associated with occipital cephaloceles in a family expressing a spectrum of phenotypes. McNiven, V et al. Am J Med Genet A. 2019 May. PMID: 30773799
- Mutations in extracellular matrix genes NID1 and LAMC1 cause autosomal dominant Dandy-Walker malformation and occipital cephaloceles. Darbro, BW et al. Hum Mutat. 2013 Aug. PMID: 23674478
Type XVIII collagen
A member of the multiplexin family, type XVIII collagen is a basement membrane trimeric protein that features collagen and proteoglycan characteristics. It is expressed as three different α chain isoforms (short, medium and long) with distinct N-terminals and a common C-terminal endostatin domain that can inhibit angiogenesis, cell migration and tumor growth. It is important for the structural maintenance of basement membranes.
Knobloch syndrome-1 (type XVIII collagen α1 chain)
Autosomal recessive disease caused by mutations in the COL18A1 gene that leads to a lack of type XVIII collagen in basement membranes, and ocular abnormalities (high myopia, retinal detachment, vitroretinal degeneration) and skull defects (occipital encephalocele). Col18a1-KO mice present with eye abnormalities, skull deformities and alterations in other tissues (brain and kidney) associated with basement membrane broadening.
See clinical phenotypes for more information
Papers
- Lack of collagen XVIII/endostatin results in eye abnormalities. Fukai, N et al. EMBO J. 2002 Apr 2. PMID: 11927538
- Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Sertie, AL et al. Hum Mol Genet. 2000 Aug 12. PMID: 10942434
- Structurally altered basement membranes and hydrocephalus in a type XVIII collagen deficient mouse line. Utriainen, A et al. Hum Mol Genet, 2004 Sep 15. PMID: 15254016